
Faster Pileup Loading with BAI Indices
23 Jan 2015One of the key features of CycleDash is the embedded pileup view, currently provided by BioDalliance. This lets you inspect the reads which contributed to a variant call:
 (This pileup shows a single nucleotide variation between the top sequence (C)
and the bottom sequence (T), relative to a reference genome (C).)
(This pileup shows a single nucleotide variation between the top sequence (C)
and the bottom sequence (T), relative to a reference genome (C).)
We noticed that the two pileup tracks were quite slow to load, often taking in excess of 30 seconds to appear. This latency discouraged use of the feature, so we set out to improve it.
Pileup data is typically stored in a BAM file, which encodes all the short reads in the order in which they appear in the genome. Because genomes are large and sequenced at high read depth, BAM files can be quite large. For example, the synth3 BAM files from the DREAM Somatic Mutation Calling Challenge weigh in at 177.4GB each.
Transferring such a large file to a user’s web browser is impractical. Instead, BioDalliance (and other genome browsers such as IGV) load a BAM Index (BAI), which they then use to load only small portions of the larger BAM file.
As BAM files grow, so do their corresponding BAI index files. For the synth3 data set, the two BAI files are each 8.6MB (4.9MB compressed). This is still quite a bit of information to transfer to a web browser, and it accounted for the slow load time that we were seeing:
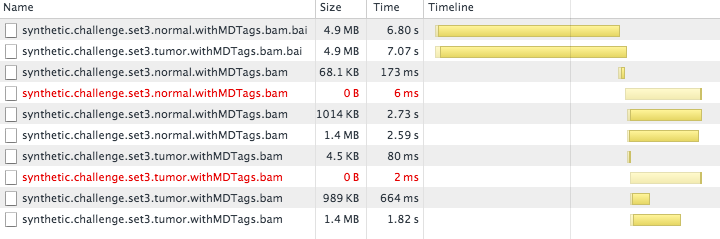
(The two BAI files block the loading of other resources for seven seconds.)
We tried some standard tricks like applying GZip compression to the BAI files, but the size of these files remained a fundamental barrier to interactive use.
In principle, only a tiny portion of the BAI file is needed at any given time. For example, if the user wants to look at Chromosome 20, then there’s no need to load index data for Chromosome 3.
After some discussion on BioStars and the samtools-devel mailing list, we decided to index the BAM index! This results in a small JSON file which records the beginning and ending of each contig in the BAI file (“contig” is a general term for a contiguous stretch of DNA, e.g. a chromosome):
{
"chunks": [
[8, 716520], // byte offsets for Chromosome 1
[716520, 1463832], // byte offsets for Chromosome 2
[1463832, 2070072], // etc.
...
]
}
We call this an “index chunk file” because it records the locations of different chunks of the BAM Index. For example, the above file records that Chromosome 3 begins at byte 1,463,832 in the BAI file and extends through byte 2,070,072. To display pileups from Chromosome 3, a genome viewer need only load these ~600K of the BAI file, rather than the full 8.6M. This results in a considerable speedup:
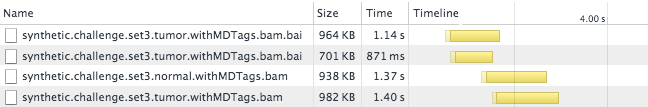
The overall load time has been reduced from 10s to something like 3s.
You might wonder, what happens if this index of the index gets too large? For a human genome, it’s quite small (1.7KB for synth3) and can be inlined into the page or requested separately. And its size depends only on the number of contigs, not the number of reads, so it won’t grow with sequencing depth (like a BAI file does).
To incorporate BAI indices into your own software, you’ll want to look at the
bai-indexer Python project and the change to BioDalliance
which added support. We typically store the index chunks in a .bai.json file
alongside the standard .bam and .bai files, so they never get separated.
While we’d hoped not to add new files and formats to an already large family, we found that it was impossible to incorporate higher level indexing data into the BAI file itself. By using a JSON format, we stay flexible to future improvements and extensions to this scheme.
By creating a higher level index, we were able to significantly improve the load time of genomic visualizations. We’d love to see wider support for this convention, so check out the bai-indexer project!