
Introducing Guacamole 0.0.0
30 Jan 2015Today we’re excited to announce the release of Guacamole, a variant calling platform that can run an order of magnitude faster than existing solutions and allows for faster iteration on variant calling algorithms.
Background
Variant calling, the process of identifying differences between two or more genomes, is an important stage in many genome analysis pipelines.
Human genomes are commonly sequenced as billions of overlapping short reads that are aligned in ways that maximize agreement about the underlying base pairs, but the data is typically noisy enough that many positions show evidence of being two or more values:
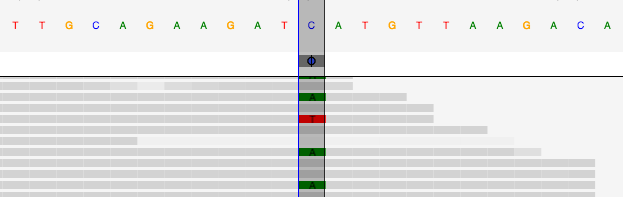
In addition, at hundreds of gigabytes of data per genome, a typical variant caller only attempts to process a few genomes and can take hours to run. This has slowed the development of better variant callers; who wants to tweak parameters and experiment on algorithms with a turn-around time of hours?
As a result, a few callers have become de facto standards, but they weren’t designed to scale to thousands (let alone millions) of genomes and, worse, there is mounting evidence that they diverge in their output (for fixed inputs), sometimes wildly!
All downstream treatment decisions hinge on the variants discovered in patients’ genomes, so it is crucial that we gain better understanding of how to quickly and accurately identify variants.
Guacamole
With this in mind, we developed Guacamole, which makes it easy to implement variant calling algorithms and run them on modern, distributed infrastructure. Along with Cycledash (our variant visualizer) and Ketrew (our workflow manager), we’re able to iterate on variant callers, run them, and view their output easily and in a matter of minutes.
What’s in 0.0.0
- Three reference variant callers, all expressed in ~100-200 lines of code and that run on full genomes in as few as 20 minutes:
- “Germline standard” caller: emits any variants whose likelihood (based on the quality of the reads displaying them) clears a certain threshold.
- “Germline threshold” caller: emits variants that appear in above a certain percentage of the reads.
- “Somatic standard” caller: compares a “tumor” sample to a “normal” sample (both having been aligned to the same reference) and emits likely variants that are specific to the former (e.g. mutations found in a cancer tumor).
- Primitives for performing genomic analysis:
partitionLociByApproximateDepth:- Assigns genomic regions to processing tasks, balancing the number of reads that align to each region.
- High coverage regions are divided into many small partitions; low coverage regions are merged.
pileupFlatmap:- Run a user-specified function on each locus of the genome, passing it a pileup giving the relevant bases from all reads mapping to that locus.
windowFlatMap:- Run a user-specified function on sliding windows of a specified size in the genome.
- All reads mapping within a window centered around each locus are passed to the user function.
- Like
pileupFlatmap, but for algorithms that require additional context around each locus.
What Are We Doing With It?
- We used Guacamole to enter variant calls into the recent DREAM Somatic Mutation Calling Challenge.
- We’ve also begun to tailor it to various clinical applications, including the development of precision cancer immunotherapies with collaborators from Mt. Sinai and Memorial Sloan Kettering.
Limitations / Roadmap
The callers included in 0.0.0 are experimental; we don’t recommend using them in production yet. Below are some things we are working on adding or improving.
Structural Variants
While the SNV-calling functionality in Guacamole is fairly robust, the indel- and SV-calling functionality is still fairly naïve.
- Better structural-variant calling is being worked on.
- We also plan to allow Guacamole to ingest reads from multiple sequencing platforms, e.g. longer PacBio reads, and make calls based on “hybrid” short- and long-read data.
Scalability
Spark, which we use as our distributed runtime, is difficult to debug and frequently fails on inputs larger than a few hundred GB (compressed; this can end up being tens of TB in deserialized-Scala-object form).
- We have built a prototype variant caller using the Crunch/Scrunch interfaces to Hadoop’s implementation of MapReduce to establish that these algorithms do complete when run on more robust runtimes.
- We are actively working on Spark’s monitoring capabilities.
- Building a pipeline capable of rapidly analyzing genomic data from hundreds or thousands of patients is a high priority.
Clinical Applications
Guacamole will be in used in various downstream applications in the coming months:
- An upcoming cancer vaccine trial will use somatic variant calls from Guacamole (in conjunction with other callers) to identify candidate biomarkers in patients’ cancer cells.
- We are studying concordance between existing variant callers, Guacamole included, in an effort to better characterize the failure modes of current approaches.
You can find more info about Guacamole, as well as see what we’re working on and file issues, on GitHub.