
Exploring the Genome with Ensembl and Python
04 Feb 2015The sequencing of the human genome took 13 years to complete, cost $3 billion dollars, was lauded as “a pinnacle of human self-knowledge”, and even inspired a book of poetry. The actual result of this project is a small collection of very long sequences (one for each chromosome), varying in length from tens to hundreds of millions of nucleotides (A’s, C’s, T’s, and G’s). For comparison, the King James Bible contains only about 3 million letters, which would make for a very diminutive chromosome.
What’s in this colossal nucleic tome? The sequence of the human genome only hints at its structure and organization. Where are the genes? Which parts are transcribed and how are they spliced? We need annotations on top of the sequence to tell us how the genome gets read.
What is Ensembl?
Realizing that sequencing the genome is only half the battle, in 1999 the European Molecular Biology Laboratory partnered with the Sanger Institute to make an annotation database called Ensembl. Ensembl catalogues where all the suspected active elements in a genome reside and to what degree they have been validated experimentally.
Many different kinds of information are gathered together within Ensembl but the most fundamental annotations are:
-
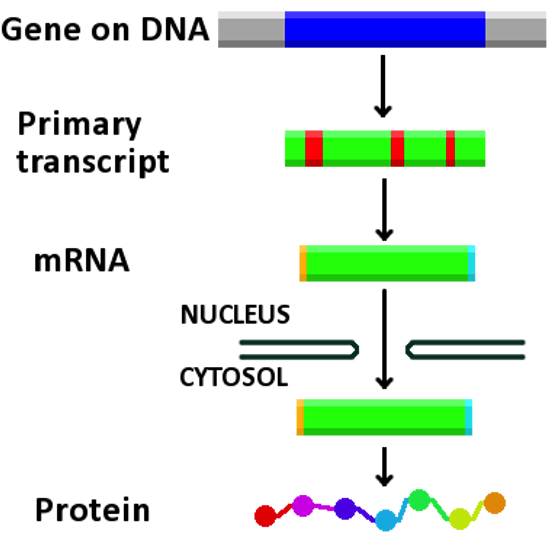
Genes: Transcriptionally active regions of the genome which may give rise to several related proteins or non-coding RNAs. A gene is characterized by a range of positions on a particular chromosome and can contain multiple transcripts.
-
Transcripts: A subset of a gene which gets copied into RNA (by a process called transcription). The subset of transcripts which are destined to become messenger RNAs (used as templates for constructing proteins) are also annotated with both their splice boundaries and the specific open reading frame that gets used for translation.
{kind=link}
How to access Ensembl
Ensembl provides a very detailed web interface, through which you can inspect all the information associated with a particular gene, transcript, genetic variant, or even the genetic locations associated with a disease. If you get tired of clicking around the Ensembl website, it’s also possible to issue programmatic queries via the Ensembl REST API.
Though convenient, using a web API restricts how many distinct queries you can issue in a small amount of time. If you want to deal with thousands of genomic entities or locations, then it would be preferable to use a local copy of Ensembl’s data. Unfortunately, local installation of Ensembl is complicated and requires an up-front decision about what subset of the data you’d like to use. Furthermore, while there is a convenient Perl API for local queries, Ensembl lacks an equivalent for Python.
Introducing PyEnsembl
To facilitate fast/local querying of Ensembl annotations from within Python we wrote PyEnsembl.
PyEnsembl lazily downloads annotation datasets from the Ensembl FTP server, which it then uses to populate a local sqlite database. You don’t have to interact with the database or its schema directly. Instead, you can use a particular snapshot of the Ensembl database by creating a pyensembl.EnsemblRelease object:
import pyensembl
ensembl = pyensembl.EnsemblRelease()
EnsemblRelease constructs queries for you through a variety of helper methods (such as locations_of_gene_name), some of which return structured objects that correspond to annotated entities (such as Gene, Transcript, and Exon).
By default, PyEnsembl will use the latest Ensembl release (currently 78). If, however, you want to switch to an older Ensembl release you can specify it as an argument to the EnsemblRelease constructor:
ensembl = pyensembl.EnsemblRelease(release=75)
Note: the first time you use a particular release, there will be a delay of several minutes while the library downloads and parses Ensembl data files.
Example: What gene is at a particular position?
We can use the helper method EnsemblRelease.genes_at_locus to determine
what genes (if any) overlap some region of a chromosome.
genes = ensembl.genes_at_locus(contig="1", position=1000000)
The parameter contig refers to which contiguous region of DNA we’re interested in (typically a chromosome). The result contained in genes is [Gene(id=ENSG00000188290, name=HES4)]. This means that the transcription factor HES4 overlaps the millionth nucleotide of chromosome 1. What are the exact boundaries of this gene? Let’s pull it out
of the list genes and inspect its location properties.
hes4 = genes[0]
hes4_location = (hes4.contig, hes4.start, hes4.end)
The value of hes4_location will be ('1', 998962, 1000172), meaning that this whole gene spans only 1,210 nucleotides of chromosome 1.
Does HES4 have any known transcripts? By looking at hes4.transcripts you’ll see that it has 4 annotated transcripts: HES4-001, HES4-002, HES4-003, and HES4-004. You can get the cDNA sequence of any transcript by accessing its sequence field. You only can also look at only the protein coding portion of a sequence by accessing the coding_sequence field of a transcript.
Example: How many genes are there in total?
To determine the total number of known genes, let’s start by getting the IDs of all the genes in Ensembl.
gene_ids = ensembl.gene_ids()
The entries of the list gene_ids are all distinct string identifiers (e.g. “ENSG00000238009”).
If you count the number of IDs, you’ll get a shockingly large number: 64,253! Aren’t there supposed to be around 22,000 genes? Where did all the extra identifiers come from?
The Ensembl definition of a gene (a transcriptionally active region) is more liberal than the common usage (which typically assumes that each gene produces some protein). To count how many protein coding genes are annotated in Ensembl, we’ll have to look at the biotype associated with each gene.
To get these biotypes, let’s first construct a list of Gene objects for each ID in gene_ids.
genes = [ensembl.gene_by_id(gene_id) for gene_id in gene_ids]
We still have a list of 64,253 elements, but now the entries look like Gene(id=ENSG00000238009, name=RP11-34P13.7). Each of these Gene objects has a biotype field, which can take on a daunting zoo of values such as “lincRNA”, “rRNA”, or “pseudogene”. For our purposes, the only value we care about is “protein_coding”.
coding_genes = [gene for gene in genes if gene.biotype == 'protein_coding']
The length of coding_genes is much more in line with our expectations: 21,983.
Limitations and Roadmap
Hopefully the two examples above give you a flavor of PyEnsembl’s expressiveness for working with genome annotations. It’s worth pointing out that PyEnsembl is a very new library and still missing significant functionality.
- PyEnsembl currently only supports human annotations. Future releases will support other species by specifying a
speciesargument toEnsemblRelease(e.g.EnsemblRelease(release=78, species="mouse")). - The first time you use PyEnsembl with a new release, the library will spend several minutes parsing its data into a sqlite table. This delay could be cut down significantly by rewriting the data parsing logic in Cython.
Transcriptobjects currently only have access to their spliced sequence but not to the sequences of their introns.- PyEnsembl currently only exposes core annotations (those found in Ensembl’s GTF files), but lacks many secondary annotations (such as APPRIS).
You can install PyEnsembl using pip by running pip install pyensembl. If you encounter any problems with PyEnsembl or have suggestions for improving the library, please file an issue on GitHub.